Myélome multiple
| Maladie | |||
_HE_stain.jpg) | |||
| Caractéristiques | |||
|---|---|---|---|
| Signes | Splénomégalie, Cachexie, Macroglossie, Pâleur, Ecchymoses, Hépatomégalie, Souffle cardiaque, Oedème, Tachycardie , Crépitements, ... [+] | ||
| Symptômes |
Confusion, Déshydratation, Macroglossie, Paresthésies, Malabsorption, Anorexie , Rétention urinaire, Oligurie, Nausées, Dyspnée , ... [+] | ||
| Diagnostic différentiel |
Ostéomalacie, Myélome multiple, Métastases osseuses, Macroglobulinémie de Waldenstrom, Leucémie à plasmocytes, Plasmocytome solitaire, Ostéogénèse imparfaite, Plasmocytose réactive, Ostéoporose, Gammapathie monoclonale de signification indéterminée | ||
| Informations | |||
| Terme anglais | Multiple myeloma | ||
| Autres noms | Maladie de Kahler | ||
| Wikidata ID | Q467635 | ||
| Spécialités | hématologie, oncologie | ||
| |||
Le myélome multiple (MM) est une prolifération malignes de plasmocytes provenant d'un seul clone et est caractérisé par l'augmentation anormale d'une paraprotéine monoclonale menant à l'atteinte des organes cibles. Le MM fait partie du spectre des gammapathies monoclonales.
Épidémiologie
Il est estimé qu'en 2021, 3800 canadiens seront diagnostiqués avec le MM et 1600 décéderont de cette pathologie[1]. Il survient principalement dans la population gériatrique avec un âge médian au diagnostic d'environ 70 ans. Le nombre de cas diagnostiqués chaque année devrait presque doubler en 20 ans. Il a un rapport homme-femme de 3 pour 2 et il est plus fréquent chez les Afro-Américains.[2][3]
Étiologies
L'étiologie exacte du MM est inconnue. Cependant, il existe des preuves suggérant que des anomalies génétiques oncogéniques tels que CMYC, NRAS et KRAS peuvent jouer un rôle dans le développement de la prolifération des plasmocytes. Des altérations chromosomiques peuvent aussi être retrouvées chez les patients atteints de MM, tels l'hyperdioploidie, la délétion 13q14, les translocations t(11;14)(q13;q32), t(4;14)(p16;q32), et t(14;16), l'amplification 1q ou la délétion 1p, et les délétions 17p13[4]. Le MM a également été associé à d'autres facteurs tels que la consommation d'alcool, l'obésité, des causes environnementales telles que les insecticides, les solvants organiques et l'exposition à l'irradiation.[5][3] Il est aussi plus prévalent chez les fermiers, les travailleurs du bois, les travailleurs du cuir et ceux avec une grande exposition au pétrole.[4]
Physiopathologie
Il est présumé que le MM résulte d'un stade précancéreux et asymptomatique de la croissance de plasmocytes monoclonaux appelé gammopathie monoclonale de signification indéterminée (MGUS), une gammapathie détectable chez plus de 3 % des personnes âgées de plus de 50 ans. Il semble que la cellule d'origine est un plasmocyte post-germinal. La progression clinique vers un MM se produit à un taux d'environ 1 % par an.[6][3]
Bien que les causes exactes du développement de la MGUS et de la progression vers le MM restent inconnues, les deux étapes fondamentales de la pathogenèse du MM sont : [3]
- Le développement d'un MGUS : possiblement en raison d'anomalies cytogénétiques générées lors d'une réponse anormale à un antigène, ce qui entraîne la production d'immunoglobulines monoclonales.[3]
- La progression du MGUS vers le MM : la progression est considérée comme une conséquence de lésions cytogénétiques supplémentaires acquises par le clone du plasmocyte d'origine, causées soit par une instabilité génétique, soit par des anomalies du microenvironnement hématopoïétique.[3]
Les plasmocytes malins dans le MM sont particulièrement sensibles à l'interleukine-6, qui semble être essentielle à la croissance et à la survie des tumeurs.[3]
Un excès d'immunoglobulines monoclonales peut provoquer une hyperviscosité, un dysfonctionnement plaquettaire et des lésions tubulaires rénales, entraînant respectivement des troubles neurologiques, des saignements et une insuffisance rénale. L'occupation de la moelle osseuse par le clone du plasmocyte en expansion se manifeste généralement par une anémie, une thrombocytopénie et une leucopénie.[3]
L'interaction entre les cellules de myélome et le microenvironnement osseux conduit finalement à l'activation des ostéoclastes et à la suppression des ostéoblastes, entraînant une perte osseuse puis des lésions ostéolytiques[note 1][7]. Plusieurs cascades de signalisation intracellulaire et intercellulaire, de nombreuses chimiokines et interleukines sont impliquées dans ce processus complexe.[3]
Présentation clinique
La présentation clinique du MM est hétérogène. Les signes et symptômes dépenderont du niveau d'atteinte des organes cibles. Un patient peut être asymptomatique (découverte fortuite) ou gravement malade.
Facteurs de risque
Les facteurs de risque pour le développement d'un MM sont[3][4]:
- l'âge
- l'irradiation
- l'alcool
- l'obésité
- les insecticides et les solvants organiques, être fermier
- les antédédents de MGUS
- les antécédents familiaux de MM
- les antécédents d'amyloidose
- les antécédents de plasmocytome solitaire
- l'exposition au benzène, charbon, et poussière de bois
- le sexe masculin[RR: 3:2]
- l'origine afro-américaine.
Questionnaire
La présentation clinique du MM est hétérogène, les patients peuvent être asymptomatiques et les symptômes dépendent du niveau d'atteinte des organes cibles[3][4][8]:
| Atteine | Symptômes |
|---|---|
| Symptômes constitutionnels | |
| Lésions lytiques osseuses | |
| Anémie | |
| Hypercalcémie | |
| Insuffisance rénale | |
| Syndrome d'hyperviscosité
|
|
| Amyloïdose |
Examen clinique
Les signes cliniques pouvant se présenter chez un patient avec MM sont[3][4][8]:
- les signes vitaux : fièvre (sx B, infection), tachycardie (anémie)
- l'apparence générale : pâleur (anémie), cachexie, ecchymoses (thrombocytopénie)
- l'examen ophtalmologique : hémorragie rétinienne et taches de cotton (hyperviscosité)
- l'examen ORL : macroglossie (amyloidose AL), lymphadénopathie (rare), adénopathie (plasmacytome)
- l'examen MSK : douleur osseuse, cyphose, perte de taille (compression vertébrale), faiblessedes membres inférieurs (neuropathie, compression médullaire)
- l'examen cardiovasculaire : tachycardie, souffle cardiaque éjectionnel (anémie), bruits cardiaques anormaux
- l'examen pulmonaire : diminution du murmure vésiculaire (pneumopathie restrictive due à douleur osseuse thoracique), crépitements (anasarque dû à l'insuffisance rénale)
- l'examen abdominal : hépatomégalie et splénomégalie (rare)
- l'examen des membres inférieurs : oedème (insuffisance rénale).
Il est possible qu'il n'y aille aucun signe clinique.
Examens paracliniques
CRAB (Manifestations du MM)
- hyperCalcémie
- insuffisance Rénale
- Anémie
- Bony lytic lesions (lésions lytiques osseuses)
Les examens paracliniques permettent de poser le diagnostic définitif ainsi que le pronostic (e.g.: stade R-ISS).
- une FSC : anémie, thrombocytopénie, leucopénie
- un frottis sanguin : formation en rouleaux (érythrocytes)
- une créatinine sérique : élévation de la créatinine sérique
- les électrolytes sériques : hypercalcémie
- les immunoglobulines sériques (IgA, IgG, IgM) : élévation de la protéine monoclonale
- une électrophorèse des protéines sériques : pic monoclonal (région bêta ou gamma)
- une électrophorèse des protéines urinaires : protéinurie de Bence-Jones (chaînes légères)
- l'immunofixation : détection des chaînes lourdes (A, G ou M) et des chaînes légères (kappa ou lambda)
- les chaînes légères libres sériques : élévation d'une chaîne légère (soit kappa ou lambda), ratio anormal des chaînes légères
- l'aspiration et biopsie de la moelle osseuse (avec immunohistochimie et/ou cytométrie de flux) : ≥10% de plasmocytes clonaux, plasmocytes matures, plasmocytes immatures, hypercellularité et infiltration plasmocytaire, cellules de Mott, clonalité des chaînes légères
- des études cytogénétiques (e.g.: FISH) : délétions, translocations, amplifications (e.g.: del 13, del 17p13, t(4;14) etc.) Utile pour déterminer sous-type biologique et recommendations pronostiques
- une tomodensitométrie pancorporelle à faible dose ou TEP : lésions ostéolytiques
- une IRM pancorporelle (sans contraste) : lorsque tomodensitométrie pancorporelle à faible dose ou TEP négatifs (différencier MM indolent versus actif)
- un bilan hépatique pour le suivi de la toxicité associé aux agents chimiothérapeutiques
- la LDH : élevée (facteur pronostic)
- la β2-microglobuline sérique : élévation (facteur pronostic)
-

Lésion lytique (rouge) dans l'os temporal comparé à un os normal (vert)
-

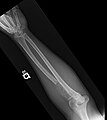
Lésions lytiques dans un humérus
-

Fracture pathologique d'une vertèbre secondaire à des lésions lytiques
-
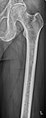
Lésions lytiques dans un fémur
-
Destruction d'un radius proximal par des lésions lytiques




Approche clinique
Lorsqu'un patient est suspecté d'avoir un MM, il est important de distinguer celui-ci des autres dyscrasies plasmocytaires à l'aide d'examens paracliniques[9]. Ils nous permettront aussi d'établir un pronostic pouvant guider l'approche thérapeutique[10][11]. Ces examens paracliniques sont décrits ci-haut et permettent d'établir le diagnostic selon le respect des critères diagnostiques décrits ci-dessous.
Il est à noter qu'il existe un chevauchement entre les signes et symptômes cliniques des dyscrasies plasmocytaires. En effet, le syndrome d'hyperviscosité est associé le plus souvent à la macroglobulinémie de Waldenström (les IgM contribuant davantage à l'augmentation de la viscosité), mais ces symptômes peuvent tout de même se présenter rarement avec un MM. D'ailleurs, l'amyloïdose peut constituer un diagnostic en soi s'il y a absence des critères diagnostiques du MM. Le MM peut rarement se développer chez les patients atteints d'amyloïdose. En contrepartie, les patients atteints de MM peuvent développer une amyloïdose superposée, qui est a suspecter lorsqu'il y a développement d'oedème, de syndrome néphrotique ou d'insuffisance cardiaque. En ce qui concerne le plasmacytome, celui-ci peut constituer un diagnostic en soi (plasmacytome isolé) s'il y a absence des critères confirmant le diagnostic de MM. La présence d'atteinte d'organe cible CRAB ou d'un ratio de chaînes légères libres > 100, de lésions lytiques à l'IRM, de >60% clones plasmocytaires médullaires confirme le diagnostic de MM chez un patient présentant un plasmocytome.[12]
Diagnostic
Pour établir le diagnostic de MM actif, les plasmocytes clonaux doivent être > 10% dans la moelle osseuse OU un plasmocytome osseux ou extramédullaire prouvé par biopsie ET au-moins un des critères suivants[13][4]:
- la présence d'un élément CRAB :
- l'hyperCalcémie: calcium sérique > 0.25 mmol/L plus haut que la limite supérieure de la normale OU > 2.75 mmol/L
- l'insuffisance Rénale: DFGe < 40 OU créatinine sérique > 177 mmol/L
- l'Anémie: hémoglobine (HB) > 20g/L en dessous de la limite inférieure de la normale OU HB < 100 g/L
- des lésions osseuses (Bony lesions):
- au moins une lésion ostéolytique sur radiographie, tomodensitométrie ou TEP
- s'il y a < 10% plasmocytes clonaux au niveau de la moelle épinière, > 1 lésion lytique osseuse est requise afin de distinguer le myélome du plasmocytome solitaire.
- la présence d'un biomarqueur indiquant une progression inévitable vers une atteinte d'un organe cible :
- 60% ou plus de plasmocytes clonaux au niveau de la moelle osseuse
- un ratio des chaînes légères (chaîne légère impliquée / chaîne légère non-impliquée) plus grand ou égal à 100 ET la chaîne légère sécrétée est quantifiée à 100 mg/L ou plus
- plus d'une lésion focale à l'IRM mesurant au moins 5 mm.
Pour établir le diagnostic de MM indolent, les deux critères diagnostiques suivants doivent être respectés[13][4]:
- Immunoglobuline monoclonale sérique (IgG ou IgA) d'au moins 30 g/L OU protéine urinaire monoclonale (protéinurie Bence-Jones) d'au moins 500mg/24h ET/OU plasmocytes clonaux de 10-60% au niveau de la moelle osseuse.
- Absence d'atteinte d'organe cible, d'anomalies biochimique (biomarqueurs tel que décrit ci-haut pour le MM actif) ou d’amyloïdose.
Diagnostic différentiel
Le diagnostic différentiel du MM inclut[3][4][8][14][15]:
- la macroglobulinémie de Waldenstrom[note 2]
- le MGUS
- le myélome multiple indolent
- les métastases osseuses d'autres cancers
- le plasmocytome solitaire[note 4][17]
- l'ostéomalacie[18] et l'ostéoporose[19][20]
- l'ostéogénèse imparfaite[19][21]
- la plasmocytose réactive (e.g.: maladies autoimmunes, cancer métastatique, VIH/SIDA, maladie hépatique chronique).
Traitement
Les patients présentant un MM symptomatique peuvent être traités par de la thérapie primaire suivie de chimiothérapique à haute dose et une greffe de cellules souches hématopoïétiques chez ceux éligibles.[22][4][11]
Traitement du MM actif
Greffe
L'éligibilité du patient pour une greffe est déterminé par son âge, ses comorbidités, et son niveau fonctionnel. La greffe est classiquement recommandée chez les patients âgés de ≤ 65 ans.[23]
| Type de greffe | Commentaires |
|---|---|
| greffe de cellules souches hématopoïétiques autologue | Après thérapie primaire d'induction, c'est l'option de traitement la plus optimale. Une greffe répétée est considérée pour le traitement d'un MM progressif ou réfractaire après thérapie primaire chez les patients ayant eu une réponse prolongée à la première greffe.[22] |
| greffe de cellules souches hématopoïétiques allogénique | Permet d'éviter la contamination engendrée par le prélèvement de cellules tumorales autologue. Il y a aussi avantage de par l'effet greffon versus tumeur associé à la greffe allogénique. Cette thérapie est cependant limitée par l'âge très avancé et les multiples comorbidités des patients atteints de myélome, ainsi que la difficulté de trouver un donneur adéquat et compatible.[22] |
Les agents myélotoxiques (ex.: agents alkylants, melphalan) sont à éviter chez les patients en attente de greffe.
Chimiothérapie
Des régimes de traitement à 3 agents sont préférés, démontrant un meilleur taux de réponse et un plus haut taux de survie sans progression (progression free survival). Des régimes à 2 agents sont utilisés lorsque le patient présente de multiple comorbidités, un bas niveau de performance/autonomie et s'il n'est pas candidat à la greffe. Un 3e agent peut être ajouté lorsque le niveau de performance du patient évolue favorablement.[12]
| Protocole | Détails | Commentaires |
|---|---|---|
| CyBorD[24] |
|
|
| VRd[25] |
|
|
| KRd[26] | Cycle 1:
Cycles 2 à 12:
Cycles 13 à 18:
Cycles 19 et au-delà (Rd):
Le régime carfilzomib/lenalidomide/dexaméthasone présente une efficacité similaire aux deux thérapies précédentes mais un risque de toxicité cardiaque, pulmonaire et rénale non-négligeable. |
|
| Rd[27] |
|
|
| IRd[28] |
|
|
| DRd[29] | Cycles 1 et 2:
Cycles 3 à 6:
Cycles 7 et au-delà:
|
|
Chez les patients éligibles et non-éligibles à la greffe, des agents de maintenance avec le lenalidomide, le bortézomib, l'ixazomib ou une combinaison de bortézomib/lenalidomine +/- dexaméthasone peuvent être utilisés.[22]
Les rechutes peuvent être traitées avec plusieurs combinaisons des mêmes agents mentionnés ci-haut, ainsi que d'autres régimes incluant la bendamustine, l'isatuximab, le panobinostat, le pomalidomide et l'elotuzumab.[22]
Traitements de support
Les traitements de support doivent également être offerts aux patients avec MM. Ces traitements visent surtout à pallier les complications et atteintes d'organes cibles dues au MM[22].
Pour tous[30]:
- vaccin contre le pneumocoque
- vaccin contre le zona
- vaccin contre l'influenza
- vaccin contre la Covid-19.
| Complication(s) | Traitement / Conduite |
|---|---|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
La thromboprophylaxie est indiquée selon le risque thromboembolique (le score IMPEDE[36] permet de stratifier le risque thromboembolique chez les patients atteints de MM):
|
Traitement du MM indolent
Le traitement du MM indolent dépendra de la stratification du risque selon l'échelle décrite dans la section évolution. Les patients à bas risque peuvent être suivis à des intervalles de 3 à 6 mois. Les patients à haut risque peuvent être inscrits à des essais cliniques, observés à des intervalles de 3 mois ou traités avec lenalidomide selon leur profil et la politique de certains centres.[12]
Suivi
Le suivi se fait selon des investigations périodiques visant à déterminer l'évolution de la maladie, ses complications, ainsi que la réponse au traitement.
Les investigations suivantes sont indiquées dans le suivi du MM actif[22]:
- un bilan de base incluant un bilan hépatique, un calcium, une albumine et une β2-microglobuline sérique
- les immunoglobulines sériques, une électrophorèse des protéines sériques, une électrophorèse des protéines urinaires, l'immunofixation, le ratio de chaînes légères libres et les chaînes légères sériques
- un TEP, une tomodensitométrie à basse dose pancorporelle ou une IRM pour le suivi radiologique au besoin (favoriser même modalité d'imagerie que celle effectuée lors du diagnostic)
- l'aspiration et biopsie de la moelle osseuse au besoin avec cytométrie de flux permettant la détection d'une maladie minime résiduelle.
Les patients asymptomatiques et ceux ayant reçu des traitements devraient avoir un suivi aux 1 à 3 mois[37].
Selon le International Response Criteria, les patients sous thérapie devraient avoir un suivi à chaque mois pour la première année, puis aux 2 mois par la suite[38].
Les investigations décrites ci-haut permettent aussi le suivi du MM indolent chez les patients à bas et à haut risque. Le suivi radiologique peut toutefois être effectué annuellement ou au besoin. Les patients à bas risque peuvent être suivis à des intervalles de 3 à 6 mois.[12]
L'éducation des patients est cruciale dans la prise en charge du MM. Les patients doivent recevoir des informations sur ce qu'est le MM, l'évolution de la maladie, les options de traitements et de soins palliatifs, ainsi que les complications et les signes et symptômes d'alarme nécessitant une consultation à l'urgence (e.g. syndrome de la queue de cheval, neutropénie fébrile).[39]
Complications
Les complications du MM sont[3][4][8][15]:
- l'hypercalcémie
- l'insuffisance rénale chronique et le syndrome néphrotique (protéinurie de Bence-Jones)
- l'immunosuppression
- les lésions squelletiques, les fractures, l'écrasement vertébral et la compression médullaire
- le syndrome d'hyperviscosité
- la neuropathie
- l'anémie
- la thrombocytopénie
- la leucopénie
- l'amyloidose (co-morbide).
Évolution
La gammapathie monoclonale de signification indéterminée (MGUS) a un risque de progression vers le MM d'environ 1 % par an.
Le MM latent, présente un risque de progression beaucoup plus élevé de 10 % par an.[3]
Un modèle de stratification révisé du risque de progression du MM latent vers l'actif propose 3 facteurs de risque[40]:
- plus de 20% de plasmocytes clonaux dans la moelle osseuse
- un taux de protéine monoclonale de plus de 20 g/L
- un ratio de chaînes légères libres au-delà de 20.
Comme pour le MGUS, la stratification du risque de progression se fait selon la présence de ces facteurs de risque (critères Mayo 20/2/20 2018)[40][22]:
| Niveau | Facteurs
de risques |
Taux de progression
à 2 ans (%) |
Temps ad progression
(mois) |
|---|---|---|---|
| Bas risque | 0 | 5 | 110 |
| Risque intermédiaire | 1 | 17 | 68 |
| Risque élevé | ≥2 | 46 | 29 |
Il existe plusieurs échelles pouvant déterminer le pronostic lié au MM. Le R-ISS (Revised International Staging System) est un des scores les plus utilisés, se basant sur l'albumine sérique, le LDH, la β2-microglobuline sérique et la cytogénétique de la maladie[4][11][10][41]:
| Critères | Stade | Survie médiane sans progression (mois) |
|---|---|---|
| β2-microglobuline < 3.5 mg/L, albumine ≥ 35 g/L avec LDH normaux sans cytogénétique à haut risque | I | 66 |
| Ne correspond pas aux stades I et III | II | 42 |
| β2-microglobuline > 5.5 mg/L ET LDH augmentés ET/OU cytogénétique à haut risque | III | 29 |
Prévention
Il est impossible de prévenir l'apparition du MM.
Notes
- ↑ En ce qui concerne les lésions ostécondensentes, elles sont presque toujours absentes dans le MM vu qu'il y a inhibition ostéblastique. Des cas de MM avec des lésions ostéocondensantes ont toutefois été rapportés.
- ↑ * IgM monoclonal.
- Tableau clinique d'anémie et de syndrome d'hyperviscosité.
- Pas de CRAB.
- ↑ *> 10% de plasmocytes à la biopsie de la moelle osseuse (de novo ou MM connu)
- > 20% de plasmocytes au niveau du sang périphérique
- Cytopénies prononcées
- Lésions osseuses moins fréquentes
- Plasmocytes absolus plus de 2 x 10^9 / L
- ↑ Tumeur de cellules plasmatiques (idem au MM). Peut être divisé en plasmacytome solitaire des os, plasmacytome extramédullaire, plasmacytomes solitaires multiples. Pas d'évidence d'atteinte systémique (pas de CRAB). Moins de 10% de plasmocytes clonaux au niveau de la moelle osseuse. Peut progresser vers le MM (approx. 2-4 ans)
Références
- Cette page a été modifiée ou créée le 2021/09/30 à partir de Multiple Myeloma (StatPearls / Multiple Myeloma (2021/07/30)), écrite par les contributeurs de StatPearls et partagée sous la licence CC-BY 4.0 international (jusqu'au 2022-12-08). Le contenu original est disponible à https://www.ncbi.nlm.nih.gov/pubmed/30521185 (livre).
- ↑ (en) « Multiple myeloma statistics », sur cancer.ca, (consulté le 6 novembre 2021)
- ↑ María-Victoria Mateos et Ola Landgren, « MGUS and Smoldering Multiple Myeloma: Diagnosis and Epidemiology », Cancer Treatment and Research, vol. 169, , p. 3–12 (ISSN 0927-3042, PMID 27696254, DOI 10.1007/978-3-319-40320-5_1, lire en ligne)
- ↑ 3,00 3,01 3,02 3,03 3,04 3,05 3,06 3,07 3,08 3,09 3,10 3,11 3,12 3,13 et 3,14 (en) « Multiple Myeloma », PubMed, 2021 jan (lire en ligne)
- ↑ 4,00 4,01 4,02 4,03 4,04 4,05 4,06 4,07 4,08 4,09 4,10 et 4,11 Munshi NC, Longo DL, Anderson KC. Plasma Cell Disorders. In: Jameson J, Fauci AS, Kasper DL, Hauser SL, Longo DL, Loscalzo J. eds. 'Harrison's Principles of Internal Medicine, 20e'. McGraw Hill; 2018. Accessed November 06, 2021. https://accessmedicine-mhmedical-com.acces.bibl.ulaval.ca/content.aspx?bookid=2129§ionid=192018291
- ↑ Madhav V. Dhodapkar, « MGUS to myeloma: a mysterious gammopathy of underexplored significance », Blood, vol. 128, no 23, , p. 2599–2606 (ISSN 1528-0020, PMID 27737890, Central PMCID 5146746, DOI 10.1182/blood-2016-09-692954, lire en ligne)
- ↑ Christoph Röllig, Stefan Knop et Martin Bornhäuser, « Multiple myeloma », Lancet (London, England), vol. 385, no 9983, , p. 2197–2208 (ISSN 1474-547X, PMID 25540889, DOI 10.1016/S0140-6736(14)60493-1, lire en ligne)
- ↑ Denis Mulleman, Cédric Gaxatte, Gaëlle Guillerm et Xavier Leroy, « Ostéocondensation diffuse révélant un myélome multiple », Revue du Rhumatisme, vol. 71, no 1, , p. 92–96 (DOI 10.1016/S1169-8330(03)00379-X, lire en ligne)
- ↑ 8,0 8,1 8,2 et 8,3 (en) Sara Mirali, Ayesh Seneviratne, Toronto Notes 2020, Toronto, , H49-H50
- ↑ 9,0 et 9,1 (en-US) Shaji K. Kumar, Natalie S. Callander, Kehinde Adekola et Larry Anderson, « Multiple Myeloma, Version 3.2021, NCCN Clinical Practice Guidelines in Oncology », Journal of the National Comprehensive Cancer Network, vol. 18, no 12, , p. 1685–1717 (ISSN 1540-1405 et 1540-1413, DOI 10.6004/jnccn.2020.0057, lire en ligne)
- ↑ 10,0 et 10,1 (en) « Revised Multiple Myeloma International Staging System (R-ISS) », sur MDCalc (consulté le 7 novembre 2021)
- ↑ 11,0 11,1 et 11,2 Delage, Robert et Cloutier, Stéphanie. Notes de cours: système hématopoiétique, cahier 2, partie 2 : Les cancers hématopoiétiques (Hiver 2020).
- ↑ 12,0 12,1 12,2 12,3 12,4 12,5 12,6 12,7 12,8 et 12,9 (en) « NCCN Clinical Practice Guidelines in Oncology: Multiple Myeloma », sur NCCN, (consulté le 18 juillet 2022)
- ↑ 13,0 et 13,1 (en-US) « International Myeloma Working Group (IMWG) criteria for the diagnosis of multiple myeloma », sur International Myeloma Foundation (consulté le 7 novembre 2021)
- ↑ (en) « Multiple myeloma differential diagnosis - wikidoc », sur www.wikidoc.org (consulté le 6 novembre 2021)
- ↑ 15,0 et 15,1 Konrad C. Nau et William D. Lewis, « Multiple Myeloma: Diagnosis and Treatment », American Family Physician, vol. 78, no 7, , p. 853–859 (ISSN 0002-838X et 1532-0650, lire en ligne)
- ↑ Michael Tveden Gundesen, Thomas Lund, Hanne E. H. Moeller et Niels Abildgaard, « Plasma Cell Leukemia: Definition, Presentation, and Treatment », Current Oncology Reports, vol. 21, no 1, , p. 8 (ISSN 1523-3790, PMID 30689121, Central PMCID 6349791, DOI 10.1007/s11912-019-0754-x, lire en ligne)
- ↑ Qurrat Ul Ain Iqbal et Haroon Javaid Majid, StatPearls, StatPearls Publishing, (PMID 34424649, lire en ligne)
- ↑ Lance Zimmerman et Brett McKeon, StatPearls, StatPearls Publishing, (PMID 31869080, lire en ligne)
- ↑ 19,0 et 19,1 (en) « Multiple myeloma differential diagnosis - wikidoc », sur www.wikidoc.org (consulté le 5 décembre 2021)
- ↑ Joann L. Porter et Matthew Varacallo, StatPearls, StatPearls Publishing, (PMID 28722930, lire en ligne)
- ↑ (en) « Osteogenesis Imperfecta - Pediatrics », sur Merck Manuals Professional Edition (consulté le 5 décembre 2021)
- ↑ 22,0 22,1 22,2 22,3 22,4 22,5 22,6 et 22,7 (en) « NCCN Clinical Practice Guidelines in Oncology: Multiple Myeloma », sur NCCN, (consulté le 13 novembre 2021)
- ↑ (en) « Which patient groups are candidates for autologous stem cell transplant to treat multiple myeloma (MM)? », sur www.medscape.com (consulté le 14 novembre 2021)
- ↑ (en) « https://www.cancercareontario.ca/en/drugformulary/regimens/monograph/46446 », sur Cancer Care Ontario (consulté le 6 décembre 2021)
- ↑ (en) « https://www.cancercareontario.ca/en/drugformulary/regimens/monograph/47476 », sur Cancer Care Ontario (consulté le 6 décembre 2021)
- ↑ (en) « https://www.cancercareontario.ca/en/drugformulary/regimens/monograph/47766 », sur Cancer Care Ontario (consulté le 28 juin 2022)
- ↑ (en) « https://www.cancercareontario.ca/en/drugformulary/regimens/monograph/46651 », sur Cancer Care Ontario (consulté le 6 décembre 2021)
- ↑ (en) « https://www.cancercareontario.ca/en/drugformulary/regimens/monograph/48026 », sur Cancer Care Ontario (consulté le 6 décembre 2021)
- ↑ (en) « https://www.cancercareontario.ca/en/drugformulary/regimens/monograph/48036 », sur Cancer Care Ontario (consulté le 6 décembre 2021)
- ↑ « Dyscrasies plasmocytaires - Myélome multiple », sur lanthiermed.com (consulté le 17 juillet 2022)
- ↑ (en) « https://www.cancercareontario.ca/en/drugformulary/drugs/monograph/43886 », sur Cancer Care Ontario (consulté le 5 décembre 2021)
- ↑ 32,0 et 32,1 (en) « https://www.cancercareontario.ca/en/drugformulary/drugs/monograph/44246 », sur Cancer Care Ontario (consulté le 5 décembre 2021)
- ↑ « Miacalcin (calcitonin salmon) dosing, indications, interactions, adverse effects, and more », sur reference.medscape.com (consulté le 6 décembre 2021)
- ↑ « UpToDate », sur www.uptodate.com (consulté le 6 décembre 2021)
- ↑ « UpToDate », sur www.uptodate.com (consulté le 6 décembre 2021)
- ↑ Fahrettin Covut, Ramsha Ahmed, Sanchit Chawla et Frank Ricaurte, « Validation of the IMPEDE VTE score for prediction of venous thromboembolism in multiple myeloma: a retrospective cohort study », British Journal of Haematology, vol. 193, no 6, , p. 1213–1219 (ISSN 1365-2141, PMID 33997961, DOI 10.1111/bjh.17505, lire en ligne)
- ↑ (en) « International Myeloma Working Group Consensus Statement for the Management, Treatment, and Supportive Care of Patients With Myeloma Not Eligible for Standard Autologous Stem-Cell Transplantation », sur https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3918540/, (consulté le 28 juin 2022)
- ↑ (en) « MULTIPLE MYELOMA », sur albertahealthservices.ca, (consulté le 6 juin 2022)
- ↑ « UpToDate », sur www.uptodate.com (consulté le 18 juillet 2022)
- ↑ 40,0 et 40,1 Arjun Lakshman, S. Vincent Rajkumar, Francis K. Buadi et Moritz Binder, « Risk stratification of smoldering multiple myeloma incorporating revised IMWG diagnostic criteria », Blood Cancer Journal, vol. 8, no 6, , p. 59 (ISSN 2044-5385, PMID 29895887, Central PMCID 5997745, DOI 10.1038/s41408-018-0077-4, lire en ligne)
- ↑ Antonio Palumbo, Hervé Avet-Loiseau, Stefania Oliva et Henk M. Lokhorst, « Revised International Staging System for Multiple Myeloma: A Report From International Myeloma Working Group », Journal of Clinical Oncology, vol. 33, no 26, , p. 2863–2869 (ISSN 0732-183X, PMID 26240224, Central PMCID 4846284, DOI 10.1200/JCO.2015.61.2267, lire en ligne)